Retour à la page du CRMR Marhea
Maladie de Fabry
 La maladie de Fabry est une affection héréditaire rare, qui toucherait environ une personne sur 40.000. Elle résulte d’un déficit en alpha-galactosidase A, une enzyme dont l’absence entraîne l’accumulation, dans les cellules, de graisses anormales, les glycosphingolipides (GL3). Ces graisses s’accumulent dans les lysosomes (petits organites impliqués dans le métabolisme cellulaire). La maladie de Fabry fait donc partie des maladies de surcharge lysosomale. La surcharge des cellules endothéliales (cellules bordant la lumière des vaisseaux) des nerfs, des petits vaisseaux de la peau, du tube digestif, du rein, du cœur, et du cerveau, explique les principaux signes de la maladie qui apparaissent dans l’enfance ou chez l’adulte jeune. Depuis que la maladie est mieux connue la fréquence des signes chez les jeunes enfants est de plus en plus reconnue.
La maladie de Fabry est une affection héréditaire rare, qui toucherait environ une personne sur 40.000. Elle résulte d’un déficit en alpha-galactosidase A, une enzyme dont l’absence entraîne l’accumulation, dans les cellules, de graisses anormales, les glycosphingolipides (GL3). Ces graisses s’accumulent dans les lysosomes (petits organites impliqués dans le métabolisme cellulaire). La maladie de Fabry fait donc partie des maladies de surcharge lysosomale. La surcharge des cellules endothéliales (cellules bordant la lumière des vaisseaux) des nerfs, des petits vaisseaux de la peau, du tube digestif, du rein, du cœur, et du cerveau, explique les principaux signes de la maladie qui apparaissent dans l’enfance ou chez l’adulte jeune. Depuis que la maladie est mieux connue la fréquence des signes chez les jeunes enfants est de plus en plus reconnue.
Les douleurs, à type de brûlures ou de décharges électriques marquent souvent le début de la maladie, apparaissent à l’effort, à la chaleur, lors d’une fièvre. Elles touchent les extrémités, durent quelques minutes à quelques heures, sont souvent invalidantes, et entraînent une limitation des activités sportives et sociales. Plusieurs médicaments (carbamazépine) donnés préventivement permettent habituellement de diminuer l’intensité et la fréquence de ces douleurs.
Les signes digestifs peuvent comporter douleurs abdominales, nausées, vomissements, diarrhée ou constipation et peuvent être d’interprétation difficile chez l’enfant.
Les signes cutanés sont fréquents sous forme de petits points de quelques millimètres de diamètre, rouge foncés, en tête d’épingles (= angiokératomes). Ils siègent sur les fesses, les bourses, la face interne des cuisses selon une topographie en « caleçon ». Certains patients ne transpirent pas ou transpirent peu (hypohidrose). Parfois des œdèmes des jambes apparaissent.
Les manifestations oculaires n’ont aucune traduction clinique, mais sont présentes dans 90 % des cas. Elles sont un moyen de diagnostic chez l’homme et de dépistage chez la femme hétérozygote. Il s’agit d’opacités de la cornée, visibles uniquement à la lampe à fente. Parfois une cataracte se développe.
L’atteinte rénale est constante mais plus tardive et longtemps silencieuse, nécessitant un dépistage systématique. Elle se manifeste par la présence anormale de protéine dans les urines (albuminurie) en général peu abondante (0,5 à 2 g /24 heures). L’hématurie (présence de sang dans les urines) est rare. La présence des signes rénaux conduit à faire pratiquer une biopsie rénale qui permet de porter le diagnostic devant la présence de sphingolipides dans différentes cellules du rein. L’insuffisance rénale est plus tardive, s’accompagnant d’hypertension artérielle. Elle peut conduire à la dialyse (en moyenne vers l’âge de 40 ans). La transplantation rénale est efficace mais ne règle pas le problème de l’accumulation de GL3 dans d’autres organes. Plus récemment la présence de kystes dans les reins a été reconnue en échographie.
Les manifestations cardiovasculaires : l’accumulation de sphingolipides dans le cœur est responsable d’une augmentation de son épaisseur. Des anomalies de la conduction électrique et du rythme cardiaque peuvent être dépistés par un électrocardiogramme. L’obstruction des artères du cœur peut conduire à un infarctus du myocarde. Parfois une baisse de la pression artérielle au passage de la position assise à la position debout survient (hypotension orthostatique) se manifestant par des malaises et devant conduire à adopter un lever très progressif.
L’atteinte cérébrale. L’accumulation dans les petits vaisseaux cérébraux peut diminuer le flux sanguin et être responsable de troubles de l’équilibre, de bourdonnements d’oreille (acouphènes) ou d’une surdité. Des accidents vasculaires cérébraux peuvent survenir. Le dépistage de cette atteinte peut se faire par l’IRM cérébrale et un audiogramme.
Le diagnostic de la maladie est souvent difficile et donc porté avec retard. Il repose sur le dosage de l’activité alpha-galactosidase dans les globules blancs du sang et est réalisé dans plusieurs laboratoires en France à partir d’une simple prise de sang. Cette activité est nulle ou très basse chez l’homme atteint. Par contre, chez la femme hétérozygote (voir notion de base en génétique), on peut trouver soit des taux bas (qui affirment le diagnostic), soit des taux normaux qui peuvent être trompeurs. Le dosage de GL3 dans les urines peut alors être un moyen diagnostique. L’identification d’une mutation du gène de l’alpha-galactosidase permettra d’affirmer qu’une femme est ou non hétérozygote.
Le gène est situé sur le chromosome X et la maladie se transmet selon le mode dominant lié à l’X: elle touche les sujets des deux sexes, mais est plus sévère chez les hommes (XY) qui n’ont qu’un chromosome X que chez les femmes (XX) qui en ont deux, dont l’un est sain. Les hommes transmettent la mutation à toutes leur filles en général moins gravement atteintes, qui transmettent elles même une maladie plus sévère à 50% de leurs garçons. Ils ne la transmettent jamais à leur fils (qui reçoivent le chromosome Y de leur père). Les femmes transmettent la mutation à 50% de leurs enfants, filles ou garçons. Comme dans beaucoup de maladies génétiques liées à l’X (par exemple le syndrome d’Alport) les femmes hétérozygotes ne sont en général pas simplement transmettrices de la maladie, elles peuvent développer des symptômes, parfois sévères.
La recherche de mutations dans le gène codant l’alpha-galactosidase A est maintenant réalisée dans plusieurs laboratoires en France. Plus de 350 mutations différentes ont été identifiées dans le monde. Quasiment chaque famille a une mutation spécifique.
La mise à la disposition des patients d’un traitement substitutif sous forme d’ « enzyme-médicament » depuis quelques années a été bien sûr un formidable pas en avant dans le traitement de la maladie de Fabry. Deux spécialités sont disponibles en Europe, produites par génie génétique. Elles sont toutes deux administrées par perfusion intraveineuse lente toutes les 2 semaines. Le traitement est généralement bien toléré mais des réactions « allergiques » peuvent se développer. Ils peuvent correspondre au développement d’anticorps contre l’enzyme, reconnue comme étrangère par l’organisme, qui doivent être recherchés systématiquement.
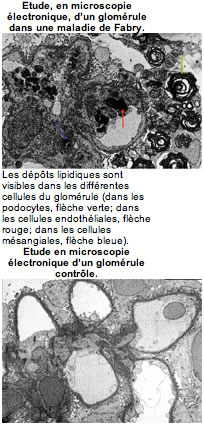
L’efficacité sur les taux de GL3 sanguin a été démontrée. L’enzymothérapie diminue la fréquence et l’intensité des douleurs, améliore la qualité de vie et semble diminuer l’épaississement du cœur (visualisé en échographie). Dans les tissus, il a été montré (en étudiant des biopsies de rein, de cœur et de peau faites avant et après traitement) que les dépôts régressent dans certaines cellules et ont tendance à persister dans d’autres, par exemple dans les podocytes (voir notions de bases sur le rein normal ). L’efficacité sur la fonction rénale à long terme reste à être évaluée. La question de la date de la mise en route du traitement reste également à préciser.
Le(s) gène(s) en cause
![]()





