Retour à la page du CRMR Marhea
Cystinurie-lysinurie classique
La cystinurie est l’une des principales causes de lithiase (c’est-à-dire de calcul) rénal héréditaire. Même si le nom de cette maladie ressemble à cystinose, ces deux pathologies n’ont rien à voir.
Génétique et mécanismes responsables de la maladies
La cystinurie est une maladie héréditaire dont le mode de transmission est autosomique récessif (voir notion de génétique). Deux gènes peuvent être en cause : soit SLC3A1 sur le chromosome 2 soit SLC7A9 sur le chromosome 19, qui codent tous deux des protéines qui sont des transporteurs présents dans les cellules du tubule rénal (et du tube digestif) et qui transportent la cystine et les acides aminés dit di-basiques (cystine, lysine, ornithine, arginine) depuis les tubules rénaux (ou le tube digestif), vers le sang. Comme la cystine est très peu soluble dans l’urine, son élimination excessive aboutit à la formation de calculs, qui sont la seule conséquence pathologique de ce trouble (l’élimination excessive des autres acides aminés n’entraîne pas de symptôme). La maladie est due à la présence d’une mutation sur chacune des deux copies (d’origine maternelle et d’origine paternelle) de l’un de ces gènes. Lorsque les 2 mutations affectent le gène SLC3A1, la cystinurie est dite de type A ; si c’est le gène SLC7A9, on parle de type B.
Les parents hétérozygotes (c’est à dire porteurs d’une mutation sur une seule des deux copie du gène) ont une cystinurie normale dans la cystinurie de type A. En revanche dans le type B, la cystinurie peut être augmentée chez les parents hétérozygotes, et ils peuvent présenter des calculs. Le diagnostic du type de mutation par l’étude de l’ADN des patients par les techniques de biologie moléculaire, qui peut être réalisé en France soit dans le laboratoire de biochimie de l’Hôpital Robert Debré à Paris soit dans le laboratoire de biochimie du CHU de Rouen est utile pour conduire l’enquête familiale, en particulier dans la fratrie des sujets atteints. En effet la sévérité de la maladie (âge de survenue des calculs, sévérité de la maladie lithiasique) peut être très variable d’un patient à l’autre, même dans une même famille. C’est la raison pour laquelle, même en l’absence de diagnostic moléculaire, un dosage de cystinurie et une échographie rénale doivent être réalisés chez tous les membres de la fratrie, même asymptomatiques, afin de mettre en route, le cas échéant, un traitement préventif de la lithiase.
Diagnostic et évolution

Les calculs de cystine sont révélés avant 20 ans dans plus de 80% des cas, et doivent être recherchés devant tout calcul du sujet jeune, surtout si les calculs sont multiples, bilatéraux ou récidivants. La cystinurie est responsable de 5 à 10 % des lithiases de l’enfant. Les calculs de cystine (figure 1) sont peu radio-opaques (c’est-à-dire sont d’un blanc assez pâle sur les radios d’abdomen), mais toujours bien visibles en échographie.
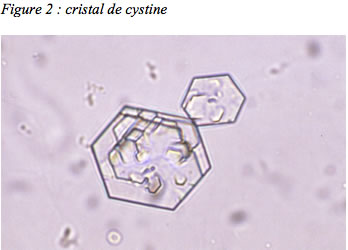
Chez les nourrissons, il n’est pas rare que de petits calculs soient éliminés spontanément et retrouvés dans les couches. Tout calcul émis doit être conservé pour être analysé : l’analyse en spectrophotométrie infrarouge permettra d’affirmer la nature cystinique du calcul. En l’absence de calcul analysable, une étude microscopique des urines fraîches du matin à la recherche de micro-cristaux (cristallurie) permet souvent d’affirmer le diagnostic en mettant en évidence les cristaux hexagonaux de cystine, tout-à-fait reconnaissables ( figure 2).
L’excrétion quotidienne (par 24h) normale de cystine ne dépasse pas 0,13 millimole (mmol) (soit 31 mg) par jour chez l’adulte (à corriger par la « surface corporelle » chez l’enfant). Lorsque l’on n’a pas les urines de 24h on peut calculer sur un échantillon d’urines le rapport cystine sur créatinine, qui ne doit pas dépasser 0,03 mmol/mmol (ou 24 mg/g). Les patients atteints de cystinurie ont une excrétion urinaire supérieure à 2 mmol/jour (480 mg).
Le transporteur des acides aminés dibasiques est aussi présent dans le tube digestif, et l’on a remarqué, sur des échographies prénatales de foetus atteints, une échogénicité augmentée (aspect trop blanc à l’échographie) des parois intestinales, qui n’a pas de traduction clinique après la naissance, mais peut parfois permettre un diagnostic précoce.
La lithiase cystinique est très récidivante et peut de ce fait retentir sur la qualité de vie, en raison des crises douloureuses liées à l’obstruction des voies urinaires, des traitements chirurgicaux répétés et des contraintes du traitement médical préventif. On observe parfois un retentissement rénal secondaire à l’obstruction des voies urinaires et/ou à des épisodes infectieux associés qui pourraient, à terme, être responsable d’une insuffisance rénale.
Traitement et prévention
Le traitement chirurgical des calculs qui ne s’éliminent pas spontanément fait appel aux techniques urologiques habituelles du traitement des lithiases : lithotritie extra-corporelle (mais la cystine est assez résistante aux ondes de choc), extraction par urétéroscopie, néphrolithotomie percutanée, chirurgie « à ciel ouvert » … en fonction de chaque situation.
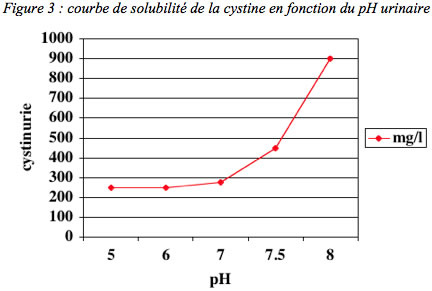 Dès le début de la prise en charge, et ensuite après disparition des calculs, un traitement médical est indispensable et permettra d’éviter les récidives, inéluctables en son absence. Le but du traitement est d’amener la concentration de cystine libre dans l’urine et le pH (taux d’acidité) urinaire au dessous du seuil de cristallisation de la cystine (figure 3), c’est-à-dire à un niveau permettant la solubilité de la cystine excrétée dans les urines. Ceci peut être obtenu théoriquement en associant plusieurs mesures diététiques et médicamenteuses.
Dès le début de la prise en charge, et ensuite après disparition des calculs, un traitement médical est indispensable et permettra d’éviter les récidives, inéluctables en son absence. Le but du traitement est d’amener la concentration de cystine libre dans l’urine et le pH (taux d’acidité) urinaire au dessous du seuil de cristallisation de la cystine (figure 3), c’est-à-dire à un niveau permettant la solubilité de la cystine excrétée dans les urines. Ceci peut être obtenu théoriquement en associant plusieurs mesures diététiques et médicamenteuses.
– le premier point et le principal est la dilution permanente des urines grâce à des boissons abondantes permettant une diurèse de l’ordre de 3 litres par 24 heures chez l’adulte (2 litres/m2 chez l’enfant). Il est d’autre part très important que ces boissons soient prises non seulement dans la journée mais également pendant la nuit, période où la concentration des urines est maximum et donc le risque de formation de lithiase également.
– d’autre part, l’alcalinisation des urines jusqu’à un pH de 7 à 7,5 est également indispensable pour obtenir la meilleure solubilité de la cystine. A un pH de 7, le seul de cristallisation de la cystine se situe environ à 250 mg/l (figure 3). Aussi, peut-on calculer le volume de la diurèse quotidienne nécessaire pour se situer toujours au dessous du seuil de cristallisation : si l’excrétion urinaire de cystine est de 750 mg/jour, une diurèse d’au moins 3 litres d’urines au pH 7 est indispensable pour éviter la précipitation de cristaux de cystine. Cette alcalinisation, qui peut être obtenue avec du Bicarbonate de Sodium ou du bicarbonate de Potassium ou avec du Citrate de Potassium doit être répartie régulièrement au cours de la journée et de la nuit. On préférera le diluer dans l’eau de boisson afin d’avoir une prise la plus régulière possible et d’améliorer la tolérance digestive.
– il est important de réduire la consommation de protides. La méthionine, précurseur de la cystine, ne peut pas être complètement supprimée de l’alimentation car il s’agit d’un acide aminé essentiel (c’est-à-dire qui ne peut pas être synthétisé par l’organisme), mais il faut limiter les apports aux besoins minimums nécessaires (environ 1200mg/jour de Méthionine). Les aliments les plus riches que sont la morue séchée, la viande de cheval, les écrevisses sont rarement consommés de façon abondante, mais les œufs, le fromage (parmesan, gruyère), la viande, le poisson, contiennent également environ 600 mg de méthionine pour 100 g et leur consommation doit être en particulier limitée au repas du soir. Limiter la consommation de viande/poisson/fromage à 120-150g /j diminue le débit de cystinurie de 0,5 à 1 mmol/j.
– il est important de réduire la consommation de sel. En effet il existe, chez les patients, une corrélation entre natriurèse (quantité de sel dans l’urine) et excrétion urinaire de cystine, sans doute liée à une réabsorption couplée des acides aminés et du sodium. Passer de 200 mmol/j (12g de NaCl) à 100 mol/l (6g de NaCl) diminue la cystinurie de 0,50 mmol/j.
– lorsque malgré toutes ces mesures, la cystinurie dépasse le seuil de cristallisation, il faut recourir à un traitement par les sulfhydriles, qui captent la cystine pour l’éliminer dans les urines. Les deux principaux médicaments de cette série sont la D-Pénicillamine (Trolovol®) et la Tiopronine (Acadione®). Seule la Tiopronine a une autorisation de mise sur le marché pour traiter la lithiase cystinique. La D-Pénicillamine est plus efficace mais moins bien tolérée par le tube digestif que la Tiopronine qui est en général mieux acceptée. Pour être efficaces, ces médicaments doivent être pris à une dose suffisante car un comprimé d’Acadione® ne peut pas complexer plus de 100 mg de cystine libre. Il faut essayer d’augmenter la dose jusqu’au niveau où l’on obtient dans les urines une concentration de cystine libre inférieure à 250 mg/l. Ces médicaments ont des effets secondaires, en particulier cutanés (éruptions, démangeaisons), digestifs, hématologiques (surveillance de la numération formule sanguine) et rénaux (surveillance régulière de la protéinurie).
Toutes ces mesures sont contraignantes mais indispensables pour prévenir autant que possible les récidives de lithiases et leurs conséquence.
Autres informations :
Il existe une association de patients atteints de cystinurie :
l’ARIC (association pour la recherche et l’information sur la cystinurie) ;
leur site internet : http://www.cystinurie.com/
Le(s) gène(s) en cause
SLC3A1
SLC7A9
![]()





