Retour à la page du CRMR Marhea
Polykystose dominante chez l’enfant
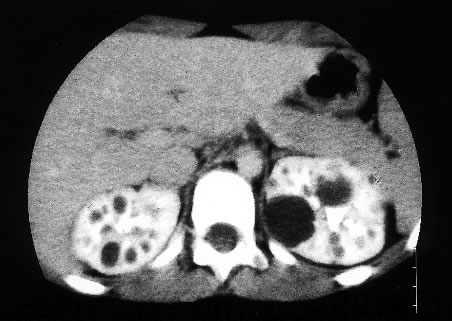 La polykystose rénale dominante (PKD) est la plus fréquente des maladies rénales héréditaires. Elle se caractérise par le développement progressif de kystes plus ou moins volumineux dans les 2 reins (voir figure), parfois associés à des kystes hépatiques, et par sa transmission autosomique dominante ; elle est associée dans 80 à 85% des cas à une mutation du gène PKD 1 situé sur le chromosome 16, ou, plus rarement, du gène PKD2 sur le chromosome 4.
La polykystose rénale dominante (PKD) est la plus fréquente des maladies rénales héréditaires. Elle se caractérise par le développement progressif de kystes plus ou moins volumineux dans les 2 reins (voir figure), parfois associés à des kystes hépatiques, et par sa transmission autosomique dominante ; elle est associée dans 80 à 85% des cas à une mutation du gène PKD 1 situé sur le chromosome 16, ou, plus rarement, du gène PKD2 sur le chromosome 4.
On l’appelle aussi « polykystose adulte » car elle se manifeste habituellement à l’âge adulte aboutissant généralement à une insuffisance rénale sévère après 50 ans. La maladie peut être découverte chez un enfant dans 3 circonstances différentes : avant la naissance sur l’échographie foetale, au cours de l’enfance ou de l’adolescence à cause d’un symptôme précoce, ou chez un enfant bien portant à l’occasion d’une échographie de dépistage ou indiquée pour une autre raison.
Avant la naissance
La PKD peut être diagnostiquée dès le 2ème ou 3ème trimestre de la grossesse lorsque l’échographie fœtale retrouve un aspect « hyperéchogène » (trop dense) des reins, qui sont également de grande taille (« gros reins hyperéchogènes »), mais on ne voit pas de vrais kystes à ce stade. Il n’est pas rare que l’échographie des parents, systématique devant un tel aspect, fasse découvrir une PKD ignorée chez l’un d’eux.
Lorsque le volume du liquide amniotique, qui témoigne du fonctionnement rénal chez le foetus, est normal, l’enfant n’aura pas de symptômes à la naissance, ni dans l’enfance dans la plupart des cas ; l’on ne sait pas encore si l’évolution vers l’insuffisance rénale de ces enfants sera plus rapide à l’âge adulte.
Dans des cas exceptionnels, la présence de très gros reins et d’une diminution importante du liquide amniotique (« oligoamnios »), témoignant d’une insuffisance rénale sévère du foetus, peut faire discuter une interruption médicale de grossesse.
Au cours de l’enfance
Hormis les cas diagnostiqués « in utero », la maladie est généralement découverte à l’occasion d’une échographie, motivée par n’importe quel symptôme (lié ou le plus souvent non lié à la polykystose) ou réalisée dans le cadre d’un dépistage familial systématique. Dans la maladie liée au gène PKD1, la plus fréquente, 50 % des enfants atteints ont déjà au moins 1 kyste visible à l’échographie à 10 ans, et ce pourcentage monte à 70 % à 20 ans (et 100% à 30 ans). Les kystes ne sont le plus souvent pas visibles chez l’enfant dans la forme associée à une mutation PKD2.
La plupart des enfants porteurs du gène muté n’ont aucun symptôme dans l’enfance, malgré le développement progressif des kystes au fil des années. Dans un faible pourcentage de cas, la maladie s’exprime, surtout à partir de 10 ans, principalement par une hypertension artérielle. La pression artérielle doit donc être mesurée régulièrement, avec un appareil adapté à l’âge, et l’hypertension doit être traitée dès qu’elle apparaît. L’insuffisance rénale, si elle n’existe pas à la naissance, n’apparaît pratiquement jamais dans l’enfance ou l’adolescence. Les autres symptômes possibles chez l’enfant sont l’hématurie (sang dans les urines), habituellement causée par un traumatisme sur un kyste ou par la présence de calculs, et parfois des douleurs abdominales ou une infection urinaire.
Les kystes du foie et les anévrysmes cérébraux ne s’observent pratiquement pas avant l’âge adulte.
Conduite à tenir
Ces enfants, qui n’ont habituellement aucun symptôme ou des symptômes bénins, doivent mener une vie normale, et peuvent en particulier pratiquer des sports, à condition d’éviter les traumatismes abdominaux. En l’absence d’hypertension artérielle, aucun traitement n’est nécessaire. L’hypertension artérielle doit être traitée dès son apparition, de préférence avec des médicaments « protecteurs rénaux » (« inhibiteurs du système rénine angiotensine » ou « sartans ») à doses adaptées à l’âge.
La surveillance porte essentiellement sur la pression artérielle (à mesurer annuellement si elle est normale) et sur l’échographie rénale ; si celle-ci est normale au premier examen, il est inutile de la répéter chaque année, un examen tous les 5 ans suffit jusqu’à 15 ans environ, âge où la procréation devient possible et où le statut de transmetteur ou non doit être connu. Chez les enfants porteurs de kystes, le rythme de surveillance dépend du volume des kystes ; habituellement, une échographie tous les 2 ou 3 ans est suffisante. De même, si la fonction rénale (mesurée par le taux de créatinine sanguine) est initialement normale, il est inutile de la vérifier tous les ans.
Quant au diagnostic génétique moléculaire (recherche de mutations dans les 2 gènes), il n’a que de rares indications; en effet les examens d’imagerie (échographie, voire scanner rénal dans les cas douteux) permettront le diagnostic en temps utile (avant la procréation) dans la grande majorité des cas.
Le(s) gène(s) en cause
- PKD1
- PKD2




