Glomérulonéphrite extramembraneuse
Qu’est-ce que la glomérulonéphrite extramembraneuse ?
Les reins contiennent des glomérules qui filtrent le sang, fonctionnant comme des passoires, en retenant les substances de grandes tailles (les protéines et les cellules sanguines) et en laissant passer les déchets, petites substances provenant surtout de l’alimentation, comme l’eau, le sel, l’urée. Ces déchets filtrés se retrouvent alors dans l’urine.
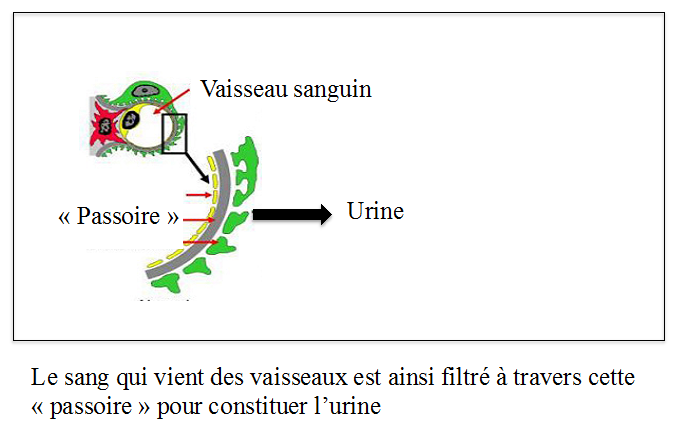 Un syndrome néphrotique survient lorsque les trous de la passoire s’élargissent laissant passer dans les urines des protéines (protéinurie), essentiellement de l’albumine (albuminurie). Il en résulte une diminution de la concentration d’albumine dans le sang qui assure normalement par un effet « éponge » le maintien de l’eau et du sel dans le sang. L’eau et le sel vont donc passer de la circulation sanguine dans les tissus et provoquer des œdèmes.
Un syndrome néphrotique survient lorsque les trous de la passoire s’élargissent laissant passer dans les urines des protéines (protéinurie), essentiellement de l’albumine (albuminurie). Il en résulte une diminution de la concentration d’albumine dans le sang qui assure normalement par un effet « éponge » le maintien de l’eau et du sel dans le sang. L’eau et le sel vont donc passer de la circulation sanguine dans les tissus et provoquer des œdèmes.
La glomérulonéphrite extramembraneuse (GEM) est la cause la plus fréquente de syndrome néphrotique de l’adulte à partir de 50 ans. Elle est caractérisée par la mise en évidence sur une biopsie du rein de dépôts granuleux d’anticorps sur le versant externe du filtre, expliquant le terme « extramembraneux ».
Le nombre exact de nouveaux cas annuels dans la population générale reste mal connu. Il est estimé à 1,3 pour 100 000 adultes. Il s’agit donc d’une maladie rare.
L’évolution est très variable d’un patient à l’autre : environ un tiers des patients entre spontanément en rémission, un tiers garde une protéinurie de débit variable et peut présenter un certain degré d’insuffisance rénale, et un tiers évolue vers une insuffisance rénale chronique sévère requérant mise en dialyse ou transplantation. La maladie peut récidiver sur le greffon après transplantation rénale dans près de 50% des cas et éventuellement compromettre sa fonction.
La GEM est parfois associée à une autre maladie comme un cancer, des infections en particulier virales (hépatite B) ou parasitaires, ou des prises de certains médicaments. Le plus souvent, elle survient sans autre maladie apparente, justifiant jusqu’à récemment le terme idiopathique.
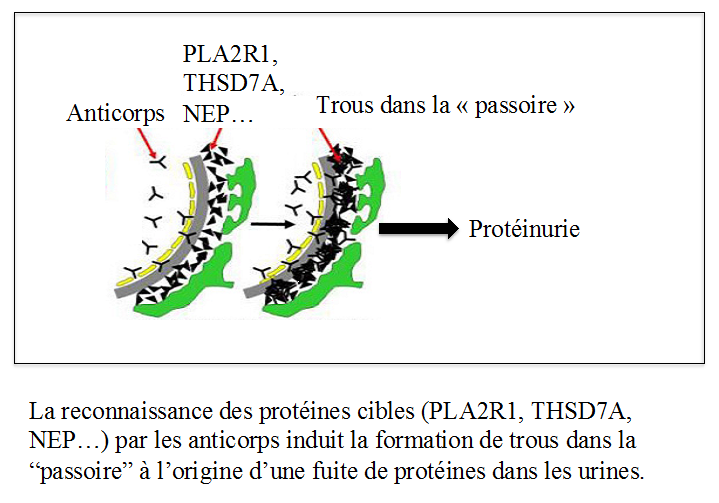 La GEM est une maladie auto-immune, caractérisée par la production d’anticorps dirigés contre nos propres antigènes localisés dans les filtres glomérulaires.
La GEM est une maladie auto-immune, caractérisée par la production d’anticorps dirigés contre nos propres antigènes localisés dans les filtres glomérulaires.
L’équipe parisienne de l’hôpital Tenon a été la première a identifier un tel antigène chez l’enfant nouveau-né (1), et a contribué avec l’équipe niçoise à identifier les antigènes chez l’adulte par différentes techniques, faisant appel à la génétique ou à la biochimie. Ces antigènes ont des noms de code dont la signification importe peu, qu’il s’agisse de NEP chez l’enfant ou chez l’adulte, de PLA2R1 dans environ 80% des cas (2), ou de THSD7A (3) dans moins de 5% des cas. Il reste donc encore d’autres antigènes à découvrir.
Notre système immunitaire fabrique normalement des anticorps pour nous protéger contre les différents pathogènes de notre environnement (bactéries, virus, parasites…). Pour des raisons encore inconnues, dans le cadre des GEM, notre organisme fabrique des anticorps contre des protéines (PLA2R1, THSD7A) qui constituent les mailles de cette passoire.
Il existe également des cas chez l’enfant qui sont dus à la production d’anticorps contre des antigènes alimentaires comme l’albumine du bœuf apportée par la viande ou le lait (4), ce qui amène à évoquer le rôle de plus en plus important de l’environnement, pollution y compris.
Un syndrome néphrotique est suspecté lorsqu’il existe des œdèmes, responsables d’une bouffissure des paupières le matin et d’un gonflement des chevilles dans la journée. L’excès d’eau et de sel dans l’organisme peut également entraîner un épanchement dans l’enveloppe (plèvre) qui entoure les poumons (épanchement pleural), et plus rarement un gonflement de l’abdomen (ascite) ou des bourses chez l’homme (hydrocèle). Ces œdèmes sont gênants, mais le plus souvent, ils ne sont pas dangereux.
Ils peuvent être facilement rattachés à un syndrome néphrotique en recherchant la présence de protéines dans les urines en utilisant une simple bandelette réactive.
Parfois, il n’y a aucun symptôme et le diagnostic est posé de façon fortuite lors d’un bilan sanguin ou urinaire qui révèle une protéinurie.
Enfin, la maladie peut être révélée par des complications diverses : douleur abdominale, essoufflement, infection, maux de tête, caillots dans la circulation (thrombose) qui peuvent se déplacer et rarement entraîner une embolie pulmonaire.
Quand le diagnostic est suspecté, on cherche à mettre en évidence les protéines dans les urines. La façon la plus simple de le faire est d’utiliser des bandelettes, type Albustix®, que l’on trempe dans les urines et dont on compare ensuite la couleur avec celles indiquées sur le flacon. La couleur de la bandelette va passer du jaune lorsqu’il n’y a pas de protéinurie au vert pâle ou vert foncé selon l’importance de la protéinurie. On peut ainsi déterminer si cette recherche de protéinurie est négative ou positive, avec une échelle à une, deux, trois ou quatre croix. La présence de « traces » ne doit pas inquiéter.
Lorsque la recherche de protéines à la bandelette est positive, il est important de préciser la quantité de protéines perdues dans les urines. Cela peut se faire au laboratoire sur un échantillon le matin ou sur un recueil d’urines durant 12 heures ou 24 heures.
Le diagnostic est ensuite confirmé par une prise de sang, recherchant notamment les anticorps spécifiques de la maladie (anti-PLA2R1 et anti-THSD7A chez l’adulte). La réalisation d’une biopsie rénale reste souhaitable chez la plupart des patients car elle apporte des éléments diagnostiques supplémentaires.
Le traitement a deux buts :
- Symptomatique, c’est-à-dire qu’il s’attaque aux symptômes (diminuer la protéinurie et les œdèmes) et vise à prévenir les complications de la maladie,
- Curatif, guérir le patient, en empêchant la production des anticorps.
Comme un tiers des patients entre spontanément en rémission, le traitement est souvent d’abord symptomatique pendant 6 mois, en l’absence de signes de gravité (insuffisance rénale progressive ou complications graves du syndrome néphrotique). Un traitement bloquant le système rénine angiotensine aldostérone (inhibiteur de l’enzyme de conversion ou bloqueur des récepteurs de l’angiotensine II) permet de diminuer la protéinurie au prix d’une baisse de pression artérielle. On y associe un régime sans sel et des diurétiques pour diminuer les œdèmes. On propose aussi dans certains cas (taux d’albumine très diminué dans le sang) des anticoagulants pour limiter le risque de thrombose et des traitements réduisant les taux de cholestérol (statine).
Le traitement curatif des GEM reste controversé. Il repose sur l’utilisation de médicaments immunosuppresseurs (qui diminuent les défenses immunitaires pour diminuer la production d’anticorps). Les corticoïdes seuls sont généralement inefficaces, et ne permettent pas d’obtenir une rémission complète c’est à dire de rendre le filtre de nouveau imperméable aux protéines et de faire disparaître la protéinurie.
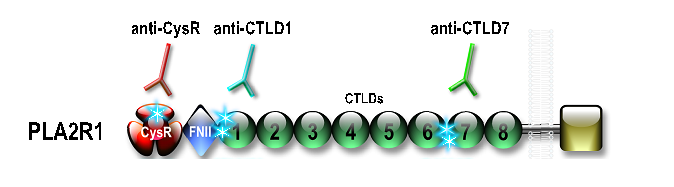
Le recours à un traitement immunosuppresseur est recommandé en cas de dégradation de la fonction rénale ou de syndrome néphrotique persistant 6 mois après l’instauration du traitement symptomatique. Cependant, ce délai d’observation peut induire des lésions irréversibles. Jusqu’à récemment, aucun marqueur ne pouvait prédire l’évolution d’un patient. La découverte des anticorps anti-PLA2R1 et anti-THSD7A a permis d’identifier de nouveaux marqueurs prédictifs de l’évolution de la fonction rénale. Un taux élevé d’anticorps anti-PLA2R1 au moment du diagnostic semble associé à une chance plus faible de rémission. Plus important que le taux élevé d’anticorps au moment du diagnostic, est sa persistance dans les semaines ou les mois qui suivent le diagnostic (5). Les anticorps anti-PLA2R1 sont dirigés contre au moins 3 domaines différents de PLA2R1. Il semble que les patients immunisés contre un seul domaine de PLA2R1 entrent plus souvent en rémission spontanée alors que les patients immunisés contre plusieurs domaines de PLA2R1 semblent présenter une évolution défavorable en l’absence de traitement curatif avec un plus fort risque d’évolution défavorable de la maladie (6,7). Le dosage des anticorps et peut-être l’analyse de leur spécificité vis-à-vis des domaines mentionnés devraient nous permettre de proposer une prise en charge plus personnalisée.
 Néanmoins, l’évolution vers l’insuffisance rénale sévère est possible nécessitant alors la mise en dialyse et/ou une greffe rénale. Dans ce cas, il existe un risque de récidive du de la GEM après transplantation rénale (8,9).
Néanmoins, l’évolution vers l’insuffisance rénale sévère est possible nécessitant alors la mise en dialyse et/ou une greffe rénale. Dans ce cas, il existe un risque de récidive du de la GEM après transplantation rénale (8,9).
Outre la surveillance régulière du taux de protéinurie et du taux sanguin d’albumine, on contrôle régulièrement la fonction rénale (urée et créatinine sanguines), et les taux d’anticorps anti-PLA2R1 (ou plus rarement anti-THSD7A). La mesure du taux des anticorps anti- PLA2R1 est disponible dans la plupart des centres hospitaliers en France. Les centres de Nice et de Paris-Tenon peuvent également réaliser la recherche d’anticorps anti-THSD7A dans le sérum.
Le centre de Nice a développé les techniques de dosage des anticorps dirigés contre certains domaines de la molécule (épitopes), qui pour l’instant sont des techniques de recherche, et dont l’utilité en clinique doit être validée et démontrée supérieure à celle du simple dosage des anticorps
1. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346(26):2053-60.
2. Beck LH, Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11-21.
3. Tomas NM, Beck LH, Jr., Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371(24):2277-87.
4. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschenes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. 2011;364(22):2101-10.
5. Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, Wakselman L, Michel PA, Mihout F, Dussol B, Matignon M, Mousson C, Simon T, Ronco P; GEMRITUX Study Group. Rituximab for Severe Membranous Nephropathy: A 6-Month Trial with Extended Follow-Up. J Am Soc Nephrol. 2017 Jan;28(1):348-358.
6. Seitz-Polski B, Dolla G, Payre C, Girard CA, Polidori J, Zorzi K, et al. Epitope Spreading of Autoantibody Response to PLA2R Associates with Poor Prognosis in Membranous Nephropathy. J Am Soc Nephrol. 2016;27(5):1517-33.
7. Seitz-Polski B, Debiec H, Rousseau A, Dahan K, Zaghrini C, Payré C, Esnault VLM, Lambeau G, Ronco P. Phospholipase A2 Receptor 1 Epitope Spreading at Baseline Predicts Reduced Likelihood of Remission of Membranous Nephropathy. J Am Soc Nephrol. 2017 Nov 7. pii: ASN.2017070734. doi: 10.1681/ASN.2017070734.
8. Debiec H, Martin L, Jouanneau C, Dautin G, Mesnard L, Rondeau E, et al. Autoantibodies specific for the phospholipase A2 receptor in recurrent and De Novo membranous nephropathy. Am J Transplant. 2011;11(10):2144-52.
9. Seitz-Polski B, Payre C, Ambrosetti D, Albano L, Cassuto-Viguier E, Berguignat M, et al. Prediction of membranous nephropathy recurrence after transplantation by monitoring of anti-PLA2R1 (M-type phospholipase A2 receptor) autoantibodies: a case series of 15 patients. Nephrol Dial Transplant. 2014;29(12):2334-42.
Rédaction : Pr Pierre RONCO (Paris – Hôpital Tenon), Pr Vincent ESNAULT (CHU Nice), Dr Barbara SEITZ-POLSKI (CHU Nice), Dr Olivia GILLION-BOYER (Paris – Hôpital Necker)




